Doença genética muito rara
Doença de Morquio A: "Aos 3 anos parei de crescer"
27/05/2024 - 12:08
Atualizado:
27/05/2024 - 12:16
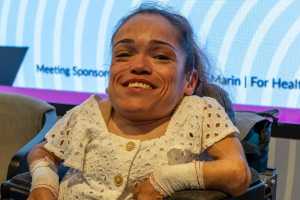
A síndrome de Morquio tipo A é uma doença hereditária do metabolismo, muito rara, progressiva e debilitante, que afeta o desenvolvimento do esqueleto. Sofia Ramos, portadora desta doença genética, revela que começou a manifestar os primeiros sintomas com 3 anos: "parei de crescer, tive algumas alterações ósseas a nível dos pulsos, tórax e quando sentada no chão não me conseguia levantar". Sem cura, o tratamento específico para a doença só está disponível em Portugal desde 2014. Trata-se de uma terapêutica de substituição enzimática que permite retardar a evolução desta patologia, à qual Sofia só teve acesso aos 25 anos de idade.
"A mucopolissacaridose (MPS) tipo IV-A, também chamada de doença de Morquio A, é uma doença genética, que resulta de um defeito no metabolismo / destruição de umas substâncias dentro organismo, denominadas de mucopolissacáridos", começa por explicar o Prof. Doutor Patrício Aguiar, professor da Faculdade de Medicina da Universidade de Lisboa e especialista em Medicina Interna na Unidade Local de Saúde do Hospital de Santa Maria, sobre esta patologia.
De acordo com o especialista, trata-se de "uma doença hereditária, na qual existe um defeito num gene denominado GALNS, que é responsável pela produção de uma enzima, a N-acetilgalactosamina-6-sulfatase". "Quando existe um defeito neste gene, a referida enzima não funciona corretamente e começa a acumular-se nas células, dentro dos lisossomas (uma espécie de fábrica de reciclagem celular), um mucopolissacárido denominado sulfato de queratano, que irá ser responsável pelas manifestações da doença", acrescenta.
O sulfato de queratano é importante para diversas funções no organismo, "mas, é essencial ao normal desenvolvimento do esqueleto, pelo que as principais manifestações desta doença ocorrem no esqueleto, com um conjunto característico de deformidades, das quais se salientam alterações da curvatura da coluna, hiperlaxidão das articulações das mãos, com mãos em garra e deformidades graves da anca e dos joelhos". Estas alterações conduzem a baixa estatura e deformação óssea.
"Além desta afetação esquelética", os doentes apresentam ainda "alterações do olho, da face, dos ouvidos, dos pulmões e das válvulas do coração e hérnias no umbigo e na região da virilha".
Os primeiros sintomas surgem, habitualmente, nos primeiros anos de vida - "geralmente, antes dos dois anos". "Os primeiros sinais estão associados às deformações do esqueleto, pelo que devemos estar particularmente atentos a alterações da curvatura da coluna (principalmente a presença de uma “giba” na porção inferior da coluna) e dos joelhos (joelhos virados para dentro). Salienta-se que em termos de altura, estes bebés nascem com uma altura normal ou até mesmo grandes e que só se começa a notar um atraso do crescimento pelos 4 a 5 anos de idade", esclarece o médico.
O seu diagnóstico passa pela avaliação da atividade da enzima N-acetilgalactosamina-6-sulfatase. "Posteriormente, pode-se ainda avaliar o gene GALNS e identificar o defeito genético específico", adianta Patrício Aguiar, sublinhando a importância do diagnóstico precoce. "Na atualidade, com a aprovação de tratamento dirigidos e específicos para doentes com MPS IV-A é essencial o diagnostico precoce, pois iniciar mais cedo a terapêutica associa-se a melhores resultados da mesma".
No entanto, admite que esta é uma área cheia de desafios. "Uma das grandes dificuldades associadas ao diagnóstico de doenças raras é a dificuldade dos médicos em conhecerem estas doenças", afirma. "Os Pediatras lidam mais frequentemente com as mucopolissacaridoses, na medida em que são doenças que se apresentam, na maioria dos casos, na infância, pelo que estão muito mais despertos para estes diagnósticos. Se pensarmos na formas menos graves de MPS IV-A, com afetação esquelética menos exuberante, o diagnóstico poderá passar despercebido até à idade adulta e os médicos de adultos dificilmente considerarão esta hipótese diagnóstica, pois raramente lidam com este tipo de doenças", justifica.
«Todos os doentes com o diagnóstico de MPS IV-A devem ser seguidos em centros de referência»
De acordo com o especialista, o tratamento para a Mucopolissacaridose tipo IV-A "divide-se entre tratamento de suporte e terapêutica específica de doença". O primeiro, explica "inclui todas as intervenções que são efetuadas que, embora não dirigidas à causa da doença em si, pretendem prevenir ou tratar as suas complicações, como, por exemplo, cirurgia de colocação de prótese da anca para o defeito esquelético da mesma". Quanto ao tratamento específico para a doença, este foi aprovado em 2014 e consiste numa terapêutica enzimática de substituição, "na qual a enzima deficitária nestes doentes é produzida em laboratório e administrada, semanalmente, por via endovenosa, aos doentes".
Segundo Patrício Aguiar, "este tratamento demonstrou melhorar a mobilidade dos doentes e estabilizar o envolvimento pulmonar pela doença". No entanto, sublinha que estes resultados "são melhores se iniciarmos o mesmo mais precocemente, pois existe menos lesão irreversível do órgãos".
Entre as principais complicações associadas, o médico destaca as dificuldades de locomoção que podem tornar o doente dependente da utilização de uma cadeira de rodas e de terceiros para realizar tarefas do dia a dia como se "vestir, lavar ou comer", uma vez que "envolvimento do sistema esquelético é grave e causa deformidades".
"Salienta-se também a exuberância das complicações respiratórias, com a necessidade de utilização de máscaras de ventilação durante o período noturno", acrescenta ainda quanto às complicações associadas.
«No meu caso aceitei bem a doença»
Apesar do diagnóstico desta patologia ter surgido "entre os 4 e 5 anos de idade", Sofia apenas iniciou o tratamento específico para a MPS IV-A aos 25 anos, após a sua aprovação em Portugal.
Embora não recorde ao certo o momento do diagnóstico, Sofia sabe que "a partir dos 3 anos" parou de crescer e começou a apresentar "algumas alterações ósseas a nível dos pulsos, tórax e quando sentada no chão não me conseguia levantar".
Sem nenhum outro caso na família, Sofia é a primeira a receber o diagnótico desta doença genética rara que lhe traz várias limitações, como falta de "mobilidade e a dificuldade em realizar tarefas diárias". Além disso, apresenta várias complicações associadas, como "complicações ósseas, respiratórias, cardíacas e auditivas". No entanto, admite que graças ao tratamento, "a doença não tem vindo a evoluir rapidamente, podendo assim dizer que a nível respiratório" se matém estável. "A nível ósseo e articular é que tenho tido mais queixas", explica referindo que após ter participado no ensaio clínico que viria a sustentar a aprovação da terapêutica em Portugal, sentiu melhorias. "Senti as mãos mais abertas, menos cansaço, mais força a nível dos membros e menos dificuldade em andar", recorda.
Não obstante, Sofia sempre soube que o tratamento não iria trazer-lhe a cura, apenas a possibilidade de a doença não progredir, melhorando a sua qualidade de vida.
Garante que sempre aceitou bem a doença, mas não pode deixar de sublinhar o desafios pessoais, sociais e até económicos associados à sua condição: "os principais desafios encontrados a nível social é a existência de preconceitos com estas pessoas que apresentam estas doenças e a não existência de inclusão na sociedade. Outra dificuldade é que não se consegue ter acesso ao mundo do trabalho e da cultura porque não existe acessibilidade. Em relação aos apoios, existem apoios, mas não são suficientes e aqueles que existem demoram para ser adquiridos. O estatuto de cuidador informal também deixa muito a desejar", lamenta.
Ainda assim, a mensagem que pretende passar é de esperança: é preciso ter força, coragem e não desistir de lutar. "Os tratamentos são importantes, não tenham receio, vale a pena fazê-los para melhorar a nossa qualidade de vida e bem-estar e principalmente ter alguma independência", afirma.
Segundo o Prof. Doutor Patrício Aguiar esta é uma doença muito rara. "Existe um estudo epidemiológico sobre a prevalência de doenças lisossomais de sobrecarga na população portuguesa, cuja publicação data do ano 2004 e que estabeleceu a prevalência de MPS tipo IV-A em 0.6 casos por cada 100.000 recém-nascidos".
"O seguimento dos doentes em centros de referência, com melhoria do tratamento de suporte e o aparecimento da terapêutica específica de doença permitiu melhorar a sobrevida destes doentes até à idade adulta e, neste momento, não sabemos exatamente qual a esperança de vida dos doentes seguidos com cuidados otimizados". Por outro lado, salienta que "a gravidade da doença é muito variável entre doentes, pelo que as formas menos graves associam-se a melhor esperança média de vida.
Autor:
Sofia Esteves dos Santos
Nota:
As informações e conselhos disponibilizados no Atlas da Saúde não substituem o parecer/opinião do seu Médico, Enfermeiro, Farmacêutico e/ou Nutricionista.
Foto:
Sofia Ramos