São doenças crónicas congénitas causadas por defeitos nos genes envolvidos no funcionamento do sistema imunitário. Até ao momento, foram identificadas mais de 350 Imunodeficiências primárias (IDP) diferentes e estima-se que possam afetar uma em cada 1200 pessoas. As IDP são condições genéticas e não estão relacionadas com a infeção pelo VIH.
As IDP apresentam uma centena de diferentes doenças, de gravidade variável. As que se manifestam no latente e na criança são geralmente mais graves que aquelas que apresentam os primeiros sintomas no adulto. Habitualmente manifestam-se por uma maior suscetibilidade a infeções, mas a desregulação do sistema imunitário pode causar o “ataque” ao próprio organismo designada doença “autoimune”.
As IDP mais frequentes são: a Deficiência seletiva de IgA, a Imunodeficiência Comum Variável, a Deficiência de subclasse de IgG e a Agamaglobulinémia ligada ao X. Entre os outros grupos de IDP destacam-se, pela sua gravidade, as IDP combinadas de linfócitos T e B e a doença granulomatosa crónica.
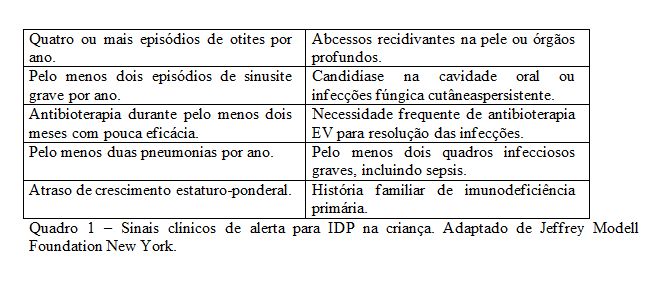
De forma simplificada deve suspeitar-se de IDP quando um doente apresentar 2 ou mais dos 10 sinais de alerta (Quadro 1).
O diagnóstico é, por vezes, difícil pois doentes com diferentes defeitos genéticos podem apresentar clínica semelhante Face à suspeita clínica, deve realizar-se exames que permitam excluir outras doenças sistémicas ou imunodeficiências secundárias mais frequentes com clínica sobreponível. A idade de apresentação, o quadro clínico, o tipo e localização da infeção e os agentes infeciosos envolvidos são determinantes na orientação do grupo de IDP a investigar. O doente deve ser orientado para uma consulta diferenciada em IDP onde são realizados exames laboratoriais criteriosamente escalonados em função do quadro clínico, incluindo o diagnóstico molecular, em que o apoio de um laboratório especializado se afigura fundamental.
É considera doença rara e deve ser sinalizada pelo médico especialista com a emissão do “cartão de doença rara”.
Cuidar de doentes com IDP é um desafio, por vezes bastante exigente, pela complexidade que algumas destas patologias podem apresentar.
Deve selecionar-se a melhor opção terapêutica para aquele caso específico, que variam deste uma abordagem terapêutica anti-infeciosa, terapêutica profilática, administração de imunoglobulina G ou transplante de células hematopoiéticas. A terapia genica afigura-se como opção terapêutica, apenas em algumas IDP monogénicas, em centros especializados internacionais.
A terapêutica de reposição com IgG polivalente é o tratamento de primeira linha nos doentes com deficiência de anticorpos e pode ser administrada mensalmente por via intravenosa em regime de hospital dia ou semanal por via subcutânea, permitindo esta a autoadministração no domicílio.
Ainda não é conhecido o risco global de infeção nos doentes com IDP, no entanto, é expectável que alguns doentes possam ter um risco maior de contrair o vírus SARS- CoV-2 ou ter uma evolução mais complicada da doença. É importante consultar o médico especialista antes de decidir tomar ou não a vacina. É recomendado que todos os doentes com IDP e função dos linfócitos T conservada sejam vacinados, especialmente aqueles com fatores de risco conhecidos para a COVID-19.
Apesar dos doentes com deficiência de anticorpos não responderem as vacinas, naqueles com a imunidade celular conservada estas vacinas podem fornecer uma proteção parcial contra a SARS- CoV-2.
É considera doença rara e deve ser sinalizada pelo médico especialista com a emissão do “cartão de doença rara”
O diagnóstico precoce e o acesso a tratamentos adequados permitem que muitos doentes com IDP tenham uma vida ativa. É fundamental a formação de todos os clínicos, no sentido de aumentar a capacidade de diagnóstico das IDP.
Aumentar o reconhecimento destas patologias e contribuir para a melhoria do acesso a cuidados médicos diferenciados são uma das prioridades do Grupo de Interesse em IDP da Sociedade Portuguesa de Alergologia e Imunologia Clínica (https://www.spaic.pt/grupos-trabalho/imunodeficiencias-primarias [1]) e da Associação Portuguesa de Doentes com IDP (https:// [2]www.adpip.org [3])

Emilia Faria MD
Assistente Graduada de Imunoalergologia
Centro Hospitalar e Universitario de Coimbra (CHUC)
Secretária-geral da SPAIC